
Dott. Ignazio Madonia
DISTROFIA MUSCOLARE E ALIMENTAZIONE
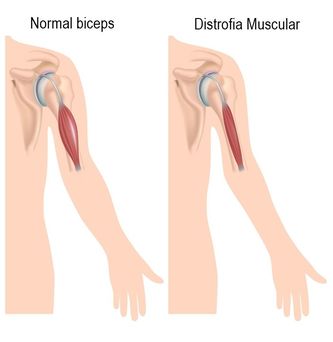
La distrofia muscolare è una malattia che affonda le sue radici nei geni.
E', infatti, a causa di un difetto nel gene che codifica per una proteina presente nelle membrane delle fibre muscolari, la distrofina, che la muscolatura di chi è affetto da questa malattia si indebolisce. La distrofina è quindi una proteina strutturale localizzata all'interno della membrana muscolare ed è legata ad altre proteine formando così un complesso proteico che ha la funzione di mantenere stabile la struttura della membrana muscolare, che viene sottoposta a continue sollecitazioni durante il ciclo di contrazione e rilasciamento tipico delle fibre muscolari.
La trasmissione di questa malattia è diaginica, cioè i maschi risultano essere sempre malati, mentre le femmine possono risultare malate nel caso in cui siano omozigoti, altrimenti sono portatrici e prive quindi di sintomi.
I due tipi principali di distrofia sono quella di Duchenne e quella di Becker.
Entrambe sono X linked ( trasmesse dai genitori alla progenie) ma a seconda del tipo di mutazione (delezione, duplicazione o mutazione puntiforme) si avranno quadri più gravi, caratterizzati dalla totale assenza di distrofina (Duchenne) oppure meno gravi, con presenza di distrofina parzialmente alterata quantitativamente o qualitativamente (Becker).
Le conseguenze sono difficoltà di movimento e di deambulazione sempre maggiori, ma non solo.
Anche il cuore e l'apparato respiratorio finiscono per essere coinvolti nel problema.
Le varie forme di distrofie si differenziano in base all'età di insorgenza, alla distribuzione del deficit muscolare e al tipo di trasmissione genetica.
Distrofia di Duchenne
Come già detto in precedenza, è una malattia X-linked che colpisce i maschi(circa 1/3500 nati) e può colpire le femmine nei rari casi in cui la madre sia portatrice e il padre malato.
La mutazione colpisce la distrofina che risulta quindi assente nelle fibre muscolari portando all'instabilità e alla necrosi delle cellule muscolari.
I 2/3 dei casi sono familiari mentre il restante 1/3 è dovuto a mutazioni sporadiche di questo gene.
Dal punto di vista clinico, alla nascita i bambini sono normali ma verso i 6 mesi – un anno si osserva il cosiddetto "floppy baby" (bambino rilasciato), bambino che non riesce a sollevarsi. È caratteristico infatti della distrofia di Duchenne il ritardo nell'apprendimento della deambulazione, tant'è che se verso i 18 mesi il bambino non è in grado di camminare scatta il sospetto di distrofia che verrà confermato dai vari esami disponibili per questa malattia.
La malattia inizia a dare i suoi segni verso il terzo anno di vita con interessamento prima del cingolo pelvico e poi di quello scapolare. Il bambino presenta difficoltà oltre che nel camminare, anche nel correre, salire le scale e cade frequentemente per l'insufficienza dei muscoli glutei.
Il decorso è rapidamente progressivo e porta alla completa incapacità di camminare verso i 10-12 anni.
L'atrofia muscolare è diffusa e l'aspetto può essere cachettico (molto magro) ma è presente, al tempo stesso, una tendenza all'obesità per l'annullamento dell’attività fisica resa impossibile dalla perdita del tessuto muscolare.
Nelle fasi successive, la malattia progredisce interessando il cuore, provocando aritmie e difetti di conduzione, mentre in fase avanzata è presente una condizione di insufficienza cardiaca che, assieme alle complicanze polmonari (insufficienza dei muscoli respiratori e fenomeni broncopneumonici) provoca la morte del paziente in genere verso i 20 anni.
Distrofia di Becker
È determinata da mutazioni dello stesso gene della distrofia di Duchenne, ma in questo caso la proteina è presente in quantità minore rispetto alla norma e presenta delle anomalie qualitative, quindi risulta essere meno stabile.
Il tipo di eredità è simile alla distrofia di Duchenne, ma meno frequente.
L'esordio più tardivo, fra i 5 e i 25 anni, con disturbi molto simili alla Duchenne ma con un'evoluzione più lenta e l'invalidità viene raggiunta anche dopo 25 anni dall'insorgenza dei sintomi.
Le lesioni cardiache sono meno frequenti e meno gravi, per cui la durata della vita può anche non subire significative variazioni: tuttavia in alcune famiglie geniche l'interessamento del cuore è molto più grave.
E', infatti, a causa di un difetto nel gene che codifica per una proteina presente nelle membrane delle fibre muscolari, la distrofina, che la muscolatura di chi è affetto da questa malattia si indebolisce. La distrofina è quindi una proteina strutturale localizzata all'interno della membrana muscolare ed è legata ad altre proteine formando così un complesso proteico che ha la funzione di mantenere stabile la struttura della membrana muscolare, che viene sottoposta a continue sollecitazioni durante il ciclo di contrazione e rilasciamento tipico delle fibre muscolari.
La trasmissione di questa malattia è diaginica, cioè i maschi risultano essere sempre malati, mentre le femmine possono risultare malate nel caso in cui siano omozigoti, altrimenti sono portatrici e prive quindi di sintomi.
I due tipi principali di distrofia sono quella di Duchenne e quella di Becker.
Entrambe sono X linked ( trasmesse dai genitori alla progenie) ma a seconda del tipo di mutazione (delezione, duplicazione o mutazione puntiforme) si avranno quadri più gravi, caratterizzati dalla totale assenza di distrofina (Duchenne) oppure meno gravi, con presenza di distrofina parzialmente alterata quantitativamente o qualitativamente (Becker).
Le conseguenze sono difficoltà di movimento e di deambulazione sempre maggiori, ma non solo.
Anche il cuore e l'apparato respiratorio finiscono per essere coinvolti nel problema.
Le varie forme di distrofie si differenziano in base all'età di insorgenza, alla distribuzione del deficit muscolare e al tipo di trasmissione genetica.
Distrofia di Duchenne
Come già detto in precedenza, è una malattia X-linked che colpisce i maschi(circa 1/3500 nati) e può colpire le femmine nei rari casi in cui la madre sia portatrice e il padre malato.
La mutazione colpisce la distrofina che risulta quindi assente nelle fibre muscolari portando all'instabilità e alla necrosi delle cellule muscolari.
I 2/3 dei casi sono familiari mentre il restante 1/3 è dovuto a mutazioni sporadiche di questo gene.
Dal punto di vista clinico, alla nascita i bambini sono normali ma verso i 6 mesi – un anno si osserva il cosiddetto "floppy baby" (bambino rilasciato), bambino che non riesce a sollevarsi. È caratteristico infatti della distrofia di Duchenne il ritardo nell'apprendimento della deambulazione, tant'è che se verso i 18 mesi il bambino non è in grado di camminare scatta il sospetto di distrofia che verrà confermato dai vari esami disponibili per questa malattia.
La malattia inizia a dare i suoi segni verso il terzo anno di vita con interessamento prima del cingolo pelvico e poi di quello scapolare. Il bambino presenta difficoltà oltre che nel camminare, anche nel correre, salire le scale e cade frequentemente per l'insufficienza dei muscoli glutei.
Il decorso è rapidamente progressivo e porta alla completa incapacità di camminare verso i 10-12 anni.
L'atrofia muscolare è diffusa e l'aspetto può essere cachettico (molto magro) ma è presente, al tempo stesso, una tendenza all'obesità per l'annullamento dell’attività fisica resa impossibile dalla perdita del tessuto muscolare.
Nelle fasi successive, la malattia progredisce interessando il cuore, provocando aritmie e difetti di conduzione, mentre in fase avanzata è presente una condizione di insufficienza cardiaca che, assieme alle complicanze polmonari (insufficienza dei muscoli respiratori e fenomeni broncopneumonici) provoca la morte del paziente in genere verso i 20 anni.
Distrofia di Becker
È determinata da mutazioni dello stesso gene della distrofia di Duchenne, ma in questo caso la proteina è presente in quantità minore rispetto alla norma e presenta delle anomalie qualitative, quindi risulta essere meno stabile.
Il tipo di eredità è simile alla distrofia di Duchenne, ma meno frequente.
L'esordio più tardivo, fra i 5 e i 25 anni, con disturbi molto simili alla Duchenne ma con un'evoluzione più lenta e l'invalidità viene raggiunta anche dopo 25 anni dall'insorgenza dei sintomi.
Le lesioni cardiache sono meno frequenti e meno gravi, per cui la durata della vita può anche non subire significative variazioni: tuttavia in alcune famiglie geniche l'interessamento del cuore è molto più grave.
Altri tipi di distrofie

- Distrofia muscolare di Emery - Dreifuss
La distrofia muscolare di Emery-Dreifuss (DMED) è caratterizzata da debolezza muscolare e atrofia, con comparsa precoce di contratture tendinee e cardiomiopatia. La prevalenza è stimata in 1/300.000. La triade clinica è costituita da 1) contratture dei tendini d'Achille, del collo e dei gomiti, a esordio nella prima infanzia, che, con il tempo, causano una limitazione della mobilità articolare; 2) atrofia e debolezza muscolare a evoluzione lenta (inizialmente con distribuzione omero-peroneale, ma in seguito più diffusa); 3) cardiopatie (difetti di conduzione, disturbi del ritmo e cardiomiopatia dilatativa) che, di solito, si manifestano dopo i 20 anni e possono provocare morte improvvisa (a volte il sintomo iniziale della malattia) e episodi ischemici da embolia.
- Distrofia muscolare congenita
Le distrofie muscolari congenite (DMC) sono un gruppo di malattie genetiche rare molto diverse l’una dall’altra, sia dal punto di vista della loro gravità ed evoluzione clinica che dal punto di vista dei meccanismi biochimici che le causano. I bambini cui si pone diagnosi di DMC hanno in comune l’esordio della debolezza muscolare alla nascita o nei primi mesi di vita (da qui il termine “congenite”), la presenza di retrazioni muscolo tendinee precoci e un quadro distrofico alla biopsia muscolare. Questi aspetti possono essere più o meno evidenti e gravi fin dall’inizio. Nelle forme più classiche si presentano infatti con il quadro del lattante ipotonico o floppy infant, a volte con prognosi molto grave per la qualità di vita, mentre in altre forme il paziente è meno compromesso dal punto di vista motorio, può stare seduto e camminare e comparire all’osservazione medica in epoca più tardiva, oltre il primo anno di vita, ma avere comunque delle caratteristiche cliniche che possono essere fatte risalire più precocemente come ad esempio una scarsa tenuta del capo o il ritardo posturo-motorio o una importante lassità legamentosa.
In alcune DMC si associano anomalie di sviluppo strutturali del cervello e del cervelletto visibili alla risonanza magnetica e possono associarsi disabilità intellettiva anche grave ed epilessia, così come problematiche cardiologiche, respiratorie, ortopediche, nutrizionali, della vista o odontoiatriche. Ne deriva che le DMC implicano l’impegno di un team multidisciplinare integrato per consentire una presa in carico adeguata dei pazienti e delle loro famiglie.
- Distrofie muscolari distali
Le distrofie muscolari distali sono un gruppo di patologie ad andamento lentamente progressivo, con coinvolgimento della muscolatura distale dei quattro arti (muscoli delle mani, degli avambracci e delle gambe). L’inizio dei sintomi avviene nell’età adulta. La distrofia muscolare di Welander esordisce con ipostenia a livello della muscolatura delle mani e progredisce lentamente con coinvolgimento della muscolatura distale degli arti inferiori. La trasmissione è autosomica dominante. La miopatia di Miyoshi, trasmessa con modalità autosomica recessiva, si manifesta inizialmente a livello della muscolatura distale degli arti inferiori e progredisce con successiva ipostenia a livello di cosce, glutei, e della muscolatura prossimale degli arti superiori. Il meccanismo patogenetico consiste in una mutazione responsabile della perdita dell’espressione della proteina disferlina.
- Distrofia facio-scapolo-omerale
La distrofia muscolare facio-scapolo-omerale (FSHD) è caratterizzata da debolezza ed atrofia muscolare. Tra le aree più colpite, i muscoli del viso (facio), le scapole (scapolo), e la parte superiore delle braccia (omerale). I segni e i sintomi della distrofia muscolare facio-scapolo-omerale di solito compaiono in adolescenza. Tuttavia, l’insorgenza e la gravità della patologia sono molto varie. In molti casi i segni della malattia possono diventare visibili in età avanzata, mentre rari casi gravi si manifestano durante l’infanzia o nella prima infanzia. La debolezza dei muscoli delle spalle può portare alla cosiddette “scapole alate”. La debolezza nei muscoli delle spalle e delle braccia può rendere difficile sollevare le braccia sopra la testa o lanciare una palla.
Fra le malattie rare, la distrofia facio-scapolo-omerale è caratterizzata da un’elevata prevalenza sulla popolazione mondiale e ha come conseguenza una significativa e progressiva perdita dell’autonomia motoria. La FSHD è la seconda forma più frequente di distrofia muscolare dell’età adulta e la terza se si considera anche l’età infantile. La prevalenza della malattia è stata stimata di 1 caso ogni 7.500/10.000 individui.
- Distrofia muscolare dei cingoli
Le distrofie muscolari dei cingoli (LGMD) sono un gruppo di malattie muscolari genetiche progressive, nelle quali è soprattutto coinvolta la muscolatura del cingolo pelvico e scapolare. Si trasmettono come carattere autosomico recessivo o dominante. Si distinguono diversi sottotipi di LGMD, in base alle analisi genetiche e proteiche. Tutte le altre malattie che presentano debolezza dei cingoli, come le sindromi dei cingoli, devono essere escluse al momento della diagnosi. Il quadro clinico della LGMD autosomica recessiva è molto simile a quello della distrofia muscolare tipo Duchenne/Becker (DMD/BMD). Nelle famiglie recessive, è molto raro l'esordio dopo i primi vent'anni, ma un esordio più tardivo è possibile nei casi dominanti. L'evoluzione della debolezza muscolare è inevitabile e può essere rapida o molto lenta. Un'indagine svolta nei Paesi Bassi, utilizzando precisi criteri diagnostici, ha identificato una prevalenza di 8,1/1.000.000 per tutti i casi di LGMD e di 5,7/1.000.000 per i casi autosomici recessivi e sporadici. E' attualmente possibile identificare diversi sottotipi di LGMD2 (forme autosomiche recessive) sulla base di mutazioni geniche e/o del deficit di prodotti genici. Non è disponibile nessun trattamento specifico; molti pazienti si sottopongono a chinesiterapia per prevenire il peggioramento delle contratture. Nella maggior parte dei pazienti, la debolezza progressiva esita in disabilità che richiede ulteriori aiuti e terapie di correzione.
- Distrofia muscolare miotonica
La Distrofia Miotonica è il tipo più comune di distrofia muscolare (atrofia o degenerazione strutturale e funzionale del tessuto muscolare) che colpisce gli individui adulti ed è caratterizzata da miopatia progressiva (alterazioni in senso anatomo-patologico, fisiologico o biochimico delle cellule o del tessuto interstiziale che compongono il muscolo), miotonia (abnorme ritardo nel rilasciamento dei muscoli volontari dopo la contrazione) e coinvolgimento multiorgano. Uno studio finlandese pubblicato sul Lancet Neurology fa il punto sulle principali caratteristiche delle due patologie evidenziando tratti comuni e differenze tra due forme della malattia.
Esistono, infatti, due forme di Distrofia Miotonica, quella di tipo 1 (anche detta ‘di Steinert’) e quella di tipo 2 (detta anche prossimale).
Entrambe le malattie sono causate da mutazioni di espansione ripetuta di nucleotidi e la trasmissione è del tipo autosomica dominante.
La Distrofia Miotonica di tipo 1 è stata descritta per la prima volta più di 100 anni fa, è una malattia muscolare atrofizzante che interessa principalmente i muscoli distali, assiali, facciali, faringei e respiratori. Determina miotonia nelle mani e danni molteplici come quelli alla cataratta, blocco della conduzione cardiaca, aritmia, diabete e sonnolenza. Questa forma è la più grave.
Nella Distrofia Miotonica di tipo 2, invece, c'è l’interessamento muscolare che prevale nei distretti prossimali degli arti (spalle, braccia, bacino e coscia). Nelle persone affette da questa forma può essere presente un’ipertrofia relativa dei muscoli della gamba (polpaccio) o un’ipotrofia asimmetrica.
- La distrofia muscolare oculo-faringea
Descritta in Italia per la prima volta nel 1975, la distrofia muscolare oculo-faringea è una malattia neuromuscolare di origine genetica, ad esordio tardivo, caratterizzata principalmente da problemi agli occhi e alla deglutizione.
Nell’ambito dei disturbi neuromuscolari, la distrofia oculo-faringea (OPMD, da Oculo-Pharyngeal Muscular Dystrophy) manifesta una relativa uniformità e tipicità dell’espressione clinica e istologica.
Ha carattere autosomico dominante – viene cioè trasmessa in linea diretta da un genitore affetto e ciascun figlio ha un rischio del 50% di ereditare la malattia – ed esordisce in genere nella quinta decade di vita, con abbassamento delle palpebre (ptosi), associato a disturbi della motilità degli occhi (oftalmoparesi) e della deglutizione. Può comparire anche debolezza degli arti, ma raramente la malattia comporta grave invalidità.
Aspetti nutrizionali

Le attuali linee guida scientifiche in termini di nutrizione rivolte a persone con malattia neuromuscolare danno delle indicazioni di base per la prevenzione e la cura della malnutrizione e dei casi di sovrappeso e obesità; da un lato viene suggerito di evitare la malnutrizione per limitare i fenomeni di sarcopenia e/o demineralizzazione ossea e, dall’altro, viene consigliato di prevenire l’eccessivo accumulo di grasso che, per caratteristica fisiologica del tessuto adiposo, si deposita anche tra le fibre muscolari andando a peggiorare il quadro istopatologico.
Tenendo conto, poi, oltre che degli aspetti puramente quantitativi (quanto mangiare), anche degli aspetti qualitativi (cosa mangiare), sono pochi gli studi sistematizzati nel campo delle malattie neuromuscolari che abbiano verificato quali alimenti sia meglio assumere. Le linee guida internazionali per la cura delle malattie neuromuscolari considerano la nutrizione come uno strumento importante per una buona salute generale e per un buon livello di qualità della vita in questi pazienti, ma non vi sono protocolli dietetici specifici da applicare in caso di malattia neuromuscolare.
Le patologie neuromuscolari presentano un aumento dello stress ossidativo (aumento dei radicali liberi) e una reazione infiammatoria esacerbata nel tessuto colpito.L’infiammazione è la prima risposta che l’organismo mette in atto contro un’infezione o un danno e ha il compito di iniziare il processo immunologico di eliminazione del patogeno e/o delle tossine e riparare, poi, il danno tissutale. E’ un meccanismo che però deve essere strettamente controllato; quando l’infiammazione si sviluppa in maniera incontrollata, essa può divenire una condizione patologica vera e propria o aggravare sintomi di malattie già esistenti nel soggetto.
Per quanto concerne lo stress ossidativo, si tratta di uno squilibrio tra la produzione di radicali liberi e le difese antiossidanti dell’organismo. I radicali sono specie reattive dell’ossigeno (ROS) e dell’azoto (RNS) prodotti dal fisiologico metabolismo cellulare e svolgono un duplice ruolo, benefico e deleterio, a seconda della loro concentrazione e localizzazione nell’organismo. Gli effetti positivi dei ROS si osservano a concentrazione bassa o moderata, queste specie sono coinvolte nella difesa nei confronti di agenti infettivi e nella trasduzione di segnali cellulari. Gli effetti dannosi di ROS e RNS si osservano quando si incorre in una modificazione nel delicato equilibro ossidanti/antiossidanti: se la generazione di ROS supera le capacità antiossidanti della cellula, o si verifica una diminuzione dei meccanismi di detossificazione, si viene ad instaurare una condizione che prende il nome di “stress ossidativo”. Lo stress ossidativo gioca un ruolo di primaria importanza in diverse patologie, comprese le malattie neuromuscolari.
La concomitante presenza di un aumento di stress ossidativo e di una reazione infiammatoria esacerbata riscontrati in soggetti con patologia NMN determinano un peggiorano il quadro istopatologico e quindi la sintomatologia.
Numerosi studi scientifici hanno dimostrato l’efficacia del Modello Alimentare Mediterraneo nel contrastare i meccanismo di stress ossidativo e di infiammazione cronica.
Con la denominazione “Dieta Mediterranea” si fa riferimento ad una gamma di diversi modelli alimentari tradizionalmente presenti nelle regioni olivicole del sud Europa e del Nord Africa, che prevede l'olio di oliva come principale fonte di grassi, un elevato consumo di verdure, frutta, legumi, cereali (preferibilmente integrali), pesce e con un consumo moderato di vino rosso durante i pasti. Rispetto alle diete standard occidentali, il Modello Alimentare Mediterraneo è caratterizzato da un quantitativo relativamente alto di acidi α-linolenico (approssimativamente 2 grammi al giorno o l’1% delle calorie totali) e un quantitativo relativamente basso di acido linoleico con un rapporto ω-3:ω-6 di 1:7.
Nella la maggior parte degli studi osservazionali e interventistici è emerso che una dieta di tipo Occidentale Standard (tipica del Nord America e Nord Europa) ricca di grassi saturi (SFA), un elevato rapporto tra acidi grassi ω- 6/ω-3, acidi grassi trans (TFA) e carboidrati ad alto indice glicemico aumenta i valori dei markers infiammatori; di contro, un tradizionale Modello Alimentare Mediterraneo, che in genere ha un elevato rapporto acidi grassi polinsaturi (PUFA) ω-3 e grassi monoinsaturi (MUFA) ω- 6 rispetto aigrassi saturi (SFA), e apporti abbondanti di frutta, verdura, legumi e cereali, mostra effetti antiinfiammatori più spiccati rispetto alle tipiche abitudini alimentari sopra riportate. I risultati ottenuti suggeriscono che il Modello Alimentare Mediterraneo è quello che meglio soddisfa le esigenze di una dieta antinfiammatoria nella pratica clinica. (NutrClinPract. 2010;25:634-640).
Il Modello Alimentare Mediterraneo, inoltre, risulta particolarmente efficace nel contrastare anche lo stress ossidativo, grazie all’apporto di alcuni costituenti degli alimenti attivi in tal senso a livello del metabolismo cellulare.
Uno studio condotto a Padova e finanziato da Telethon dimostra come si può contrastare la degenerazione muscolare grazie a un regime dietetico povero di proteine.
Poca carne: In uno studio finanziato da Telethon e pubblicato su Nature Medicine*, Paolo Bonaldo dell'Università di Padova e Marco Sandri dell'Istituto veneto di medicina molecolare e dell'Università di Padova dimostrano come promuovendo la pulizia cellulare tramite una dieta povera di proteine o certi farmaci si possano migliorare le condizioni e la forza dei muscoli distrofici.
Grazie a questo lavoro – che ha visto la collaborazione anche di altri ricercatori Telethon come Luciano Merlini dell'Università di Ferrara, Nadir Maraldi dell'Istituto Ortopedico Rizzoli di Bologna e Paolo Bernardi dell'Università di Padova – è stato dimostrato per la prima volta che si possono migliorare i sintomi della miopatia di Bethlem e della distrofia muscolare di Ullrich controllando l'autofagia, il processo fisiologico che rimuove dalle cellule sostanze tossiche oppure porzioni cellulari danneggiate.
Queste due rare malattie genetiche sono dovute a un difetto nel collagene VI, la proteina responsabile dell'ancoraggio delle fibre muscolari alla loro struttura esterna di supporto, chiamata matrice. Come già dimostrato dagli stessi ricercatori nel 2008, tra le conseguenze patologiche del difetto genetico c'è un'alterazione dei mitocondri, le "centrali energetiche" delle cellule: con la progressione della malattia i mitocondri difettosi si accumulano nelle cellule muscolari e le portano alla morte. I ricercatori Telethon hanno ora dimostrato che questi sintomi sono strettamente correlati a una inefficiente attività dell'autofagia sia nei topi distrofici sia nelle biopsie muscolari prelevate dai pazienti.
Essi hanno inoltre osservato che grazie a una dieta povera di proteine o a un trattamento farmacologico si può promuovere la "pulizia cellulare" nei topi distrofici quanto basta per rimuovere i mitocondri difettosi e mantenere le fibre muscolari pulite dalle sostanze di scarto.
Ottenendo così un miglioramento significativo della salute dei muscoli, che nel modello animale si è tradotto anche in un aumento della forza.
"L'autofagia è molto importante per un riciclo 'intelligente' delle sostanze che si accumulano nella cellula, fornisce energia quando l'apporto metabolico è insufficiente ed evita la morte cellulare quando la cellula è affollata da materiali di scarto. Poterla controllare con la dieta o con un trattamento farmacologico mirato potrebbe rivelarsi una strategia vincente per contrastare la progressione della distrofia di Ullrich e della miopatia di Bethlem".
Più in generale, il controllo dell'autofagia potrebbe contribuire a contrastare l'invecchiamento delle cellule legato all'età: consumando una dieta povera di proteine e di aminoacidi e facendo tanto movimento si può "dare una mano" ad attivare questo meccanismo e a mantenere attivo il metabolismo basale del nostro corpo. "È importante però mantenere un giusto equilibrio se l'autofagia viene attivata in modo eccessivo la cellula è portata di fatto ad 'autodigerirsi' e quindi a morire. Occorre quindi poter controllare questa attivazione: come accade generalmente in natura, il giusto equilibrio è sempre la strategia vincente".
Quindi, riassumendo la dieta deve essere orientata su alimenti antinfiammatori e antiossidanti, integrata con specifici aminoacidi come la creatina e la leucina, arricchita con vitamina D e calcio e bilanciata sia a livello energetico sia proteico, potrebbe aiutare a migliorare la qualità di vita dei pazienti, potenziando la forza e la resistenza muscolare.
Riducendo l’apporto proteico non solo si promuove la “pulizia cellulare” nei topi distrofici ma si rimuovono anche i mitocondri difettosi, mantenendo le fibre muscolari pulite dalle sostanze di scarto.
In questo modo si ottiene un netto miglioramento della salute dei muscoli, che nel modello animale si è tradotto anche in un aumento della forza. Un’alimentazione mirata potrebbe, inoltre, prevenire l’insorgere di altri rischi correlati, come l’obesità e l’osteoporosi, dovute sia alla somministrazione di terapie steroidee sia alla riduzione del movimento.
Bibliografia
*P. Grumati, L. Coletto, P. Sabatelli, M. Cescon, A. Angelin, E. Bertaggia, B. Blaauw, A. Urciuolo, T. Tiepolo, L. Merlini, N. Maraldi, P. Bernardi, M. Sandri, P. Bonaldo, “Autophagy is defective in collagen VI muscular dystrophies and its reactivation rescues myofiber de generation”. Nature Medicine, 2010
Tenendo conto, poi, oltre che degli aspetti puramente quantitativi (quanto mangiare), anche degli aspetti qualitativi (cosa mangiare), sono pochi gli studi sistematizzati nel campo delle malattie neuromuscolari che abbiano verificato quali alimenti sia meglio assumere. Le linee guida internazionali per la cura delle malattie neuromuscolari considerano la nutrizione come uno strumento importante per una buona salute generale e per un buon livello di qualità della vita in questi pazienti, ma non vi sono protocolli dietetici specifici da applicare in caso di malattia neuromuscolare.
Le patologie neuromuscolari presentano un aumento dello stress ossidativo (aumento dei radicali liberi) e una reazione infiammatoria esacerbata nel tessuto colpito.L’infiammazione è la prima risposta che l’organismo mette in atto contro un’infezione o un danno e ha il compito di iniziare il processo immunologico di eliminazione del patogeno e/o delle tossine e riparare, poi, il danno tissutale. E’ un meccanismo che però deve essere strettamente controllato; quando l’infiammazione si sviluppa in maniera incontrollata, essa può divenire una condizione patologica vera e propria o aggravare sintomi di malattie già esistenti nel soggetto.
Per quanto concerne lo stress ossidativo, si tratta di uno squilibrio tra la produzione di radicali liberi e le difese antiossidanti dell’organismo. I radicali sono specie reattive dell’ossigeno (ROS) e dell’azoto (RNS) prodotti dal fisiologico metabolismo cellulare e svolgono un duplice ruolo, benefico e deleterio, a seconda della loro concentrazione e localizzazione nell’organismo. Gli effetti positivi dei ROS si osservano a concentrazione bassa o moderata, queste specie sono coinvolte nella difesa nei confronti di agenti infettivi e nella trasduzione di segnali cellulari. Gli effetti dannosi di ROS e RNS si osservano quando si incorre in una modificazione nel delicato equilibro ossidanti/antiossidanti: se la generazione di ROS supera le capacità antiossidanti della cellula, o si verifica una diminuzione dei meccanismi di detossificazione, si viene ad instaurare una condizione che prende il nome di “stress ossidativo”. Lo stress ossidativo gioca un ruolo di primaria importanza in diverse patologie, comprese le malattie neuromuscolari.
La concomitante presenza di un aumento di stress ossidativo e di una reazione infiammatoria esacerbata riscontrati in soggetti con patologia NMN determinano un peggiorano il quadro istopatologico e quindi la sintomatologia.
Numerosi studi scientifici hanno dimostrato l’efficacia del Modello Alimentare Mediterraneo nel contrastare i meccanismo di stress ossidativo e di infiammazione cronica.
Con la denominazione “Dieta Mediterranea” si fa riferimento ad una gamma di diversi modelli alimentari tradizionalmente presenti nelle regioni olivicole del sud Europa e del Nord Africa, che prevede l'olio di oliva come principale fonte di grassi, un elevato consumo di verdure, frutta, legumi, cereali (preferibilmente integrali), pesce e con un consumo moderato di vino rosso durante i pasti. Rispetto alle diete standard occidentali, il Modello Alimentare Mediterraneo è caratterizzato da un quantitativo relativamente alto di acidi α-linolenico (approssimativamente 2 grammi al giorno o l’1% delle calorie totali) e un quantitativo relativamente basso di acido linoleico con un rapporto ω-3:ω-6 di 1:7.
Nella la maggior parte degli studi osservazionali e interventistici è emerso che una dieta di tipo Occidentale Standard (tipica del Nord America e Nord Europa) ricca di grassi saturi (SFA), un elevato rapporto tra acidi grassi ω- 6/ω-3, acidi grassi trans (TFA) e carboidrati ad alto indice glicemico aumenta i valori dei markers infiammatori; di contro, un tradizionale Modello Alimentare Mediterraneo, che in genere ha un elevato rapporto acidi grassi polinsaturi (PUFA) ω-3 e grassi monoinsaturi (MUFA) ω- 6 rispetto aigrassi saturi (SFA), e apporti abbondanti di frutta, verdura, legumi e cereali, mostra effetti antiinfiammatori più spiccati rispetto alle tipiche abitudini alimentari sopra riportate. I risultati ottenuti suggeriscono che il Modello Alimentare Mediterraneo è quello che meglio soddisfa le esigenze di una dieta antinfiammatoria nella pratica clinica. (NutrClinPract. 2010;25:634-640).
Il Modello Alimentare Mediterraneo, inoltre, risulta particolarmente efficace nel contrastare anche lo stress ossidativo, grazie all’apporto di alcuni costituenti degli alimenti attivi in tal senso a livello del metabolismo cellulare.
Uno studio condotto a Padova e finanziato da Telethon dimostra come si può contrastare la degenerazione muscolare grazie a un regime dietetico povero di proteine.
Poca carne: In uno studio finanziato da Telethon e pubblicato su Nature Medicine*, Paolo Bonaldo dell'Università di Padova e Marco Sandri dell'Istituto veneto di medicina molecolare e dell'Università di Padova dimostrano come promuovendo la pulizia cellulare tramite una dieta povera di proteine o certi farmaci si possano migliorare le condizioni e la forza dei muscoli distrofici.
Grazie a questo lavoro – che ha visto la collaborazione anche di altri ricercatori Telethon come Luciano Merlini dell'Università di Ferrara, Nadir Maraldi dell'Istituto Ortopedico Rizzoli di Bologna e Paolo Bernardi dell'Università di Padova – è stato dimostrato per la prima volta che si possono migliorare i sintomi della miopatia di Bethlem e della distrofia muscolare di Ullrich controllando l'autofagia, il processo fisiologico che rimuove dalle cellule sostanze tossiche oppure porzioni cellulari danneggiate.
Queste due rare malattie genetiche sono dovute a un difetto nel collagene VI, la proteina responsabile dell'ancoraggio delle fibre muscolari alla loro struttura esterna di supporto, chiamata matrice. Come già dimostrato dagli stessi ricercatori nel 2008, tra le conseguenze patologiche del difetto genetico c'è un'alterazione dei mitocondri, le "centrali energetiche" delle cellule: con la progressione della malattia i mitocondri difettosi si accumulano nelle cellule muscolari e le portano alla morte. I ricercatori Telethon hanno ora dimostrato che questi sintomi sono strettamente correlati a una inefficiente attività dell'autofagia sia nei topi distrofici sia nelle biopsie muscolari prelevate dai pazienti.
Essi hanno inoltre osservato che grazie a una dieta povera di proteine o a un trattamento farmacologico si può promuovere la "pulizia cellulare" nei topi distrofici quanto basta per rimuovere i mitocondri difettosi e mantenere le fibre muscolari pulite dalle sostanze di scarto.
Ottenendo così un miglioramento significativo della salute dei muscoli, che nel modello animale si è tradotto anche in un aumento della forza.
"L'autofagia è molto importante per un riciclo 'intelligente' delle sostanze che si accumulano nella cellula, fornisce energia quando l'apporto metabolico è insufficiente ed evita la morte cellulare quando la cellula è affollata da materiali di scarto. Poterla controllare con la dieta o con un trattamento farmacologico mirato potrebbe rivelarsi una strategia vincente per contrastare la progressione della distrofia di Ullrich e della miopatia di Bethlem".
Più in generale, il controllo dell'autofagia potrebbe contribuire a contrastare l'invecchiamento delle cellule legato all'età: consumando una dieta povera di proteine e di aminoacidi e facendo tanto movimento si può "dare una mano" ad attivare questo meccanismo e a mantenere attivo il metabolismo basale del nostro corpo. "È importante però mantenere un giusto equilibrio se l'autofagia viene attivata in modo eccessivo la cellula è portata di fatto ad 'autodigerirsi' e quindi a morire. Occorre quindi poter controllare questa attivazione: come accade generalmente in natura, il giusto equilibrio è sempre la strategia vincente".
Quindi, riassumendo la dieta deve essere orientata su alimenti antinfiammatori e antiossidanti, integrata con specifici aminoacidi come la creatina e la leucina, arricchita con vitamina D e calcio e bilanciata sia a livello energetico sia proteico, potrebbe aiutare a migliorare la qualità di vita dei pazienti, potenziando la forza e la resistenza muscolare.
Riducendo l’apporto proteico non solo si promuove la “pulizia cellulare” nei topi distrofici ma si rimuovono anche i mitocondri difettosi, mantenendo le fibre muscolari pulite dalle sostanze di scarto.
In questo modo si ottiene un netto miglioramento della salute dei muscoli, che nel modello animale si è tradotto anche in un aumento della forza. Un’alimentazione mirata potrebbe, inoltre, prevenire l’insorgere di altri rischi correlati, come l’obesità e l’osteoporosi, dovute sia alla somministrazione di terapie steroidee sia alla riduzione del movimento.
Bibliografia
*P. Grumati, L. Coletto, P. Sabatelli, M. Cescon, A. Angelin, E. Bertaggia, B. Blaauw, A. Urciuolo, T. Tiepolo, L. Merlini, N. Maraldi, P. Bernardi, M. Sandri, P. Bonaldo, “Autophagy is defective in collagen VI muscular dystrophies and its reactivation rescues myofiber de generation”. Nature Medicine, 2010
AVVERTENZA: Questo sito ha carattere di divulgazione culturale e informativa, necessariamente generale. Le informazioni contenute, pur basate sugli studi scientifici citati, non sostituiscono il consulto personalizzato del professionista pratico, dietologo o medico. Il lettore non è autorizzato a considerare gli articoli qui contenuti come consulti medici, né a prenderli a pretesto per curarsi da s
